Condition
Cystic Fibrosis
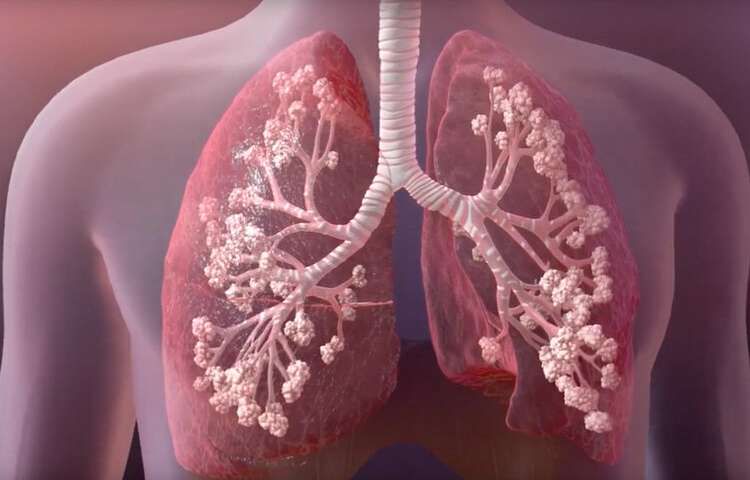
An inherited life-threatening disorder that damages the lungs and digestive system.
Cystic fibrosis affects the cells that produce mucus, sweat, and digestive juices. It causes these fluids to become thick and sticky. They then plug up tubes, ducts, and passageways.
More Research That I Did:
What Is Cystic Fibrosis?
Cystic fibrosis is a progressive, genetic disease that causes persistent lung infections and limits the ability to breathe over time.
In people with CF, mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause the CFTR protein to become dysfunctional. When the protein is not working correctly, it's unable to help move chloride -- a component of salt -- to the cell surface. Without the chloride to attract water to the cell surface, the mucus in various organs becomes thick and sticky.
In the lungs, the mucus clogs the airways and traps germs, like bacteria, leading to infections, inflammation, respiratory failure, and other complications. For this reason, minimizing contact with germs is a top concern for people with CF.
Diagnosis and Genetics:
Cystic fibrosis is a genetic disease. People with CF have inherited two copies of the defective CF gene -- one copy from each parent. Both parents must have at least one copy of the defective gene.
People with only one copy of the defective CF gene are called carriers, but they do not have the disease. Each time two CF carriers have a child, the chances are:
25 percent (1 in 4) the child will have CF
50 percent (1 in 2) the child will be a carrier but will not have CF
25 percent (1 in 4) the child will not be a carrier and will not have CF
The defective CF gene contains a slight abnormality called a mutation. There are more than 1,700 known mutations of the disease. Most genetic tests only screen for the most common CF mutations. Therefore, the test results may indicate a person who is a carrier of the CF gene is not a carrier.
Diagnosing cystic fibrosis is a multistep process, and should include a newborn screening, a sweat test, a genetic or carrier test, and a clinical evaluation at a CF Foundation-accredited care center. Although most people are diagnosed with CF by the age of 2, some are diagnosed as adults. A CF specialist can order a sweat test and recommend additional testing to confirm a CF diagnosis.
According to the Cystic Fibrosis Foundation Patient Registry, in the United States:
More than 30,000 people are living with cystic fibrosis (more than 70,000 worldwide).
Approximately 1,000 new cases of CF are diagnosed each year.
More than 75 percent of people with CF are diagnosed by age 2.
More than half of the CF population is age 18 or older.
What to Expect
Cystic fibrosis is a complex disease and the types and severity of symptoms can differ widely from person to person. Many different factors, such as age of diagnosis, can affect an individual's health and the course of the disease.
The type and severity of CF symptoms can differ widely from person to person. Therefore, although treatment plans can contain many of the same elements, they are tailored to each individual's unique circumstances.
People with CF and their families have expertise in how the disease affects them and how the context of their daily lives affect the way they approach their care. By acknowledging the respective roles, people with CF, their families, and clinical care teams can work together to develop treatment plans that align personal life goals with health goals.
The CF Foundation accredits more than 130 care centers that are staffed by dedicated healthcare professionals who provide expert CF care and specialized disease management.
Each day, people with CF complete a combination of the following therapies:
Airway clearance to help loosen and get rid of the thick mucus that can build up in the lungs.
Inhaled medicines to open the airways or thin the mucus. These are liquid medicines that are made into a mist or aerosol and then inhaled through a nebulizer and include antibiotics to fight lung infections and therapies to help keep the airways clear.
Pancreatic enzyme supplement capsules to improve the absorption of vital nutrients. These supplements are taken with every meal and most snacks. People with CF also usually take multivitamins.
An individualized fitness plan to help improve energy, lung function, and overall health
CFTR modulators to target the underlying defect in the CFTR protein. Because different mutations cause different defects in the protein, the medications that have been developed so far are effective only in people with specific mutations.
The CF Foundation supports research to discover and develop new CF treatments and maintains a pipeline of potential therapies that target the disease from every angle.
Today, the Foundation is focused on developing lifesaving new therapies for larger numbers of people with CF -- including those with rare and nonsense mutations -- and pursuing daring, new opportunities to one day develop a lifelong cure.
Research:
When a group of parents started the Cystic Fibrosis Foundation in 1955, there were no treatments for cystic fibrosis. These parents set their sights high -- to advance understanding of this little-known disease, to create new treatments and specialized care for their children, and to find a cure.
In the ensuing years, the fundraising and commitment of the CF community has enabled the Foundation to support fundamental research in the laboratory that has led to groundbreaking discoveries, including the identification of the gene and protein responsible for cystic fibrosis. By expanding our knowledge of the underlying biology of the disease and its effect on the body, researchers have paved the way for creating new treatments.
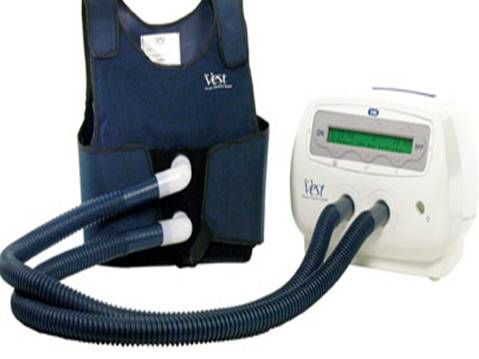
This is the vest that Cassie uses to clear up mucus in her lungs:
This is a feeding tube that patients with this disease have:
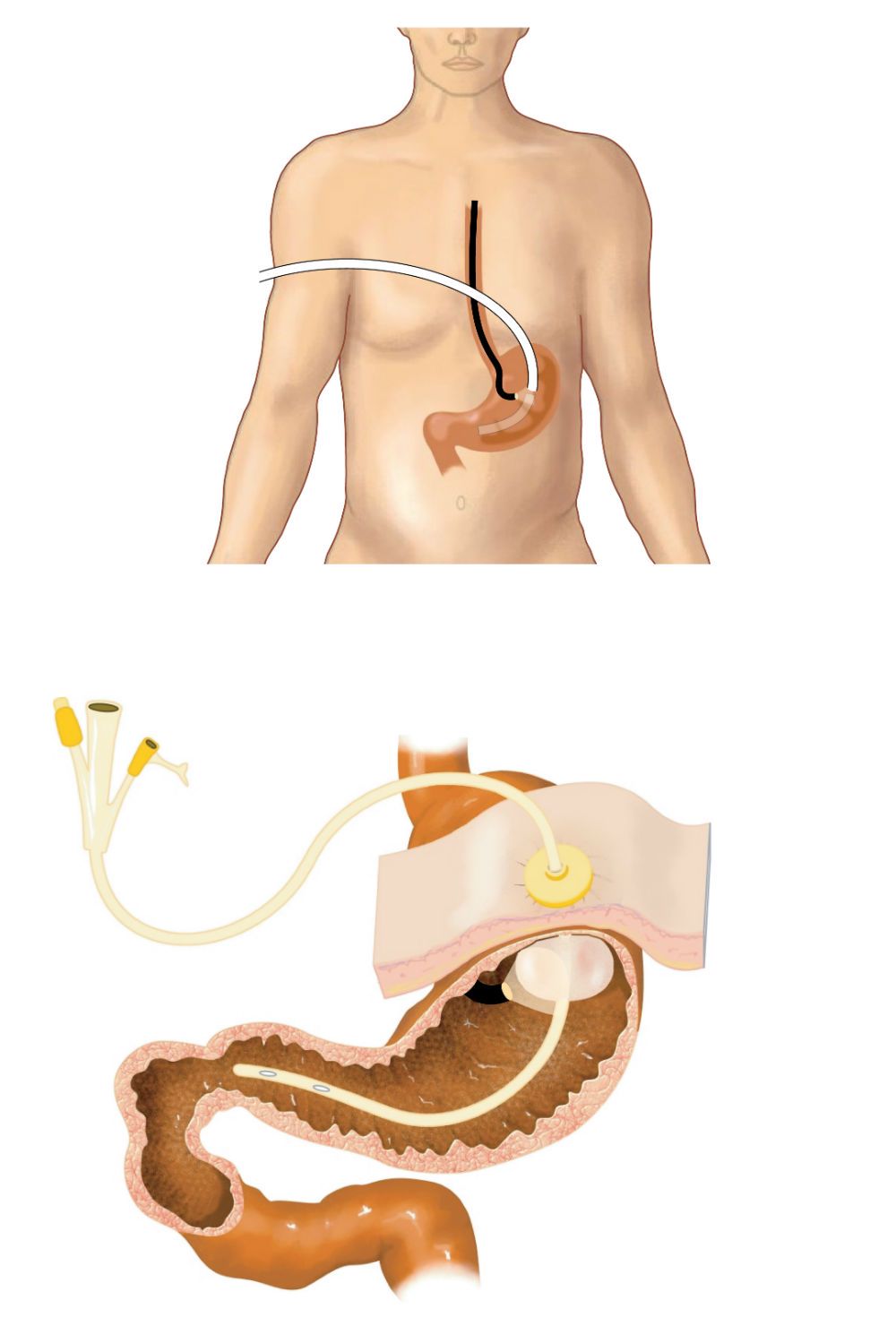
Multi-drug-resistant nontuberculous mycobacteria (NTM) can be found in the gastric juice and tubes that are used to feed some cystic fibrosis (CF) patients, research shows
Multi-drug-resistant nontuberculous mycobacteria (NTM) can be found in the gastric juice and tubes that are used to feed some cystic fibrosis (CF) patients, research shows.
Tube feeding, known as gastrostomy, involves surgeons inserting a tube through the abdominal wall and into the stomach. Gastrostomy can also be used to drain the stomach when there's a blockage.
NTM can cause many soft tissue infections. Increasingly, such infections are showing up in the lungs. NTM infection is especially worrisome in CF patients. The prevalence is as high as 32.7% of patients, depending on geographical region and patient population.
The NTM Mycobacterium abscessus complex (MABSC) is a major respiratory pathogen in CF patients. MABSC pulmonary disease in people with CF can lead to an accelerated decline in lung function and may prevent a safe lung transplant in patients with end-stage lung disease. Treatment of MABSC is not always successful and may cause adverse effects due to the multiple antibiotics that are required.
MABSC can be transmitted between CF patients despite stringent segregation. One way this can occur is through fomite contamination, or picking up bacteria through skin cells, hair, clothing or bedding in a hospital. Another way is through aerosols that CF patients generate during physiotherapy and spirometry testing.
Transmission can also occur from patients with smear-negative, culture-positive sputa, or lung fluid. This suggests that even a low level of inoculum — or material used for inoculation — could be enough for infection.
Bạn đang đọc truyện trên: AzTruyen.Top